
It is highly recommended to utilize commercial software packages ( Macromodel, Sybyl, etc.) for generating conformations.
If the user does not have access to these tools, provided Balloon software package (http://users.abo.fi/mivainio/balloon/ ) can be utilized.
1. General Settings
Balloon can accept a SMILES string (stereochemistry is recognized) or a structure that is already opened in the molecular display region.
|
|
Add Hydrogens : This option adds hydrogens if they are missing in the structure
Strip salts : Salts are removed from the structure
Keep initial structure : The initial structure is included in the output
Defaults settings : Three options are provided. The user can run with these settings but we recommend to experiment with the variables to find out the suitable conditions for the molecule
Number of maximum steps : This is the number of steps that will be tried. We recommend at least 1000 steps to sample 5 torsions.
Energy window to save conformations: Default value is 10 kcal/mol from the lowest energy conformation calculated
RMSD cutoff : If two structures atom coordinate are within 0.5 A RMSD they are assumed to be identical
2. Potential
Balloon employs a MMFF like force field for calculating the energy of conformations
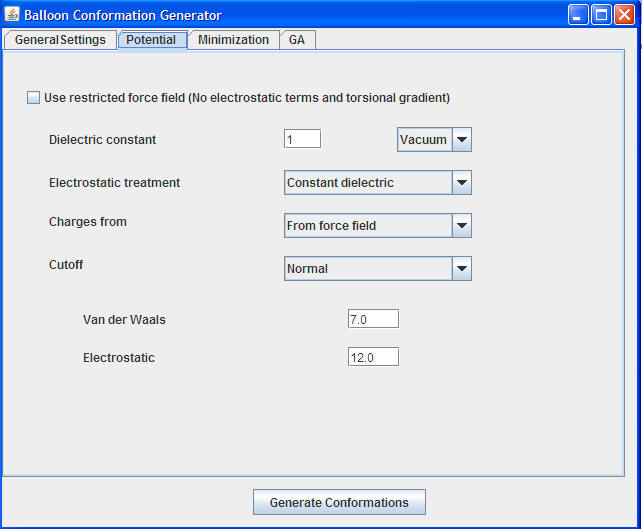
Use restricted force field: Use this option if you want to generate 3D structures very quickly. We prefer to use unrestricted force field
Dielectric Constant: Choose the dielectric constant of the solvent where the NMR experiments were conducted.
Charges from : The force field charges are good enough to generate plausible results. Refer to Balloon software documentation for more details.
Cutoff: Normal cutoff for VDW and electrostatic terms are good enough for small/mid size molecules. If the molecule is a large organic or small peptide use extended cutoffs.
3. Minimization
There is two layer of minimization. Simplex minimization is employed prior to the gradient based minimization to avoid
local minima.
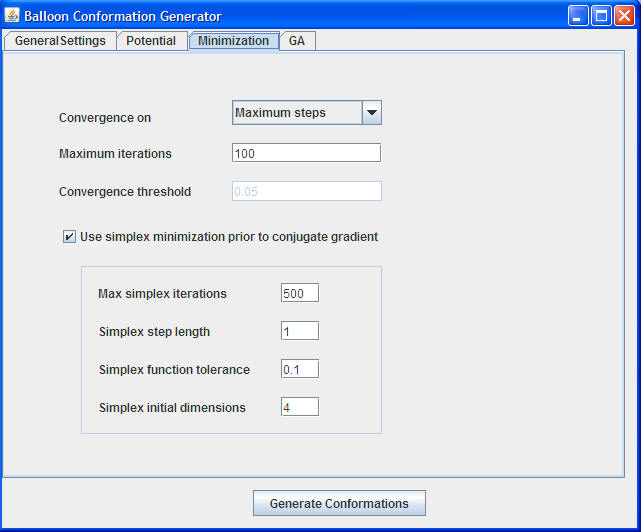
Convergence on : Maximum steps is preferred to have a linear convergence time
Maximum iterations : For small molecules 100 is enough. We prefer to set 1000-3000 steps for large molecules
Convergence threshold : 0.05 is a good threshold for most purposes
Use simplex minimization : We strongly recommend to do simplex minimizations prior to CG optimizations
Max simplex iterations : 500 is enough for most purposes. This parameter should be set to higher if the molecular size is high
Following parameters can be changed to experiment higher convergence. Please see http://users.abo.fi/mivainio/balloon/ for details.
Simplex length : Set to 1
Simplex function tolerance : Set to 0.1
Simplex initial dimensions: Set to 4.
4. Genetic Algorithm parameters
User can set to utilize distance geometry or GA for generating the conformations. If the distance geometry is chosen force field parameters will be skipped for generation of conformations.
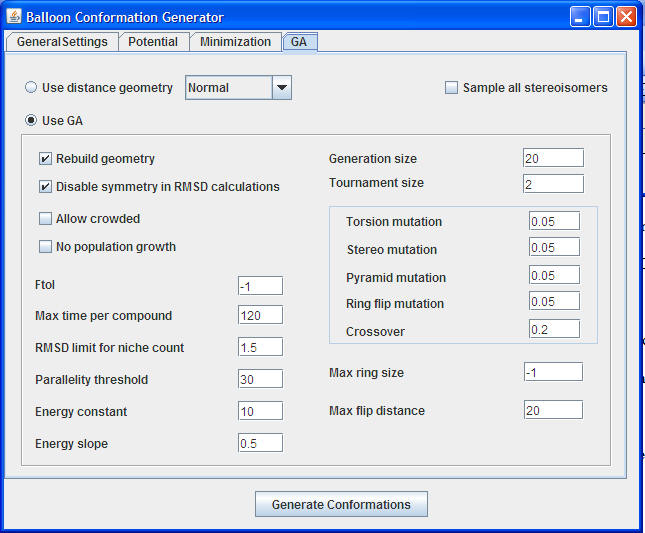
Sample all stereoisomers: If this option is selected stereochemistry of the initial conformations will be skipped
GA Settings:
Please refer to Balloon for details of the GA parameters.
http://users.abo.fi/mivainio/balloon/
| Prev | Up | Next |
| Cell Coloring | Home | DISCON Running Jobs |